
News and Business
New rules for medical devices take effect in Brazil
Learn about the main aspects of Anvisa Resolution No. 751/2022, which supersedes a previous resolution from 2001
Subjects
Effective as of March 1, 2023, the Brazilian Health Regulatory Agency’s (Anvisa) Resolution No. 751/2022 updates the rules for classifying risks, regularization procedures, labeling requirements, and preparing instructions for medical devices. The regulation fully revokes Anvisa’s Resolution No. 185/2001, which had become obsolete due to innovation in the sector over the last two decades. Although taking effect only now, the resolution was originally published in September 2022 and stems from Public Consultation No. 730/2019, which ended in January 2020 after receiving over 300 contributions.
New definitions
In adjusting Brazil’s regulations to the current reality of the industry, the provisions of Resolution No. 751/2022 feature updated definitions and new terms that are important for properly understanding how medical devices are regulated. In particular, we highlight the following:
- Medical Device: Previously defined as ‘medical product’, among other related The new definition covers any instrument, apparatus, equipment, implant, in vitro diagnostic medical device, software, material, or other article intended for use in humans for any of the following specific medical purposes:
- Diagnosing, preventing, monitoring, treating (or alleviating) illnesses and diseases;
- Diagnosing, monitoring, treating, or rehabilitating injuries or disabilities;
- Investigating, replacing, and modifying a patient’s anatomy or altering a physiological or pathological process or state;
- Providing life support or maintenance;
- Controlling or supporting conception;
- Providing information through in vitro examination of samples obtained from the human body, including organ and tissue donations;
- Aesthetic correction and cosmetic procedures, as well as for cleaning, disinfection, and sterilization.
- Medical device for in vitro diagnostics: Reagents, calibrators, standards, controls, sample collectors, software, instruments, or other articles designed for conducting in vitro examinations of samples obtained from the human body in order to obtain information to diagnose, monitor, screen, check compatibility and genetic predispositions, provide prognosis, and predict or determine a patient’s physiological This definition differs from one that was previously established in Resolution No. 36/2015).
- Software as a Medical Device (SaMD): Going into more detail than the specific regulation on the subject (Anvisa Resolution No. 657/2022), the resolution defines SaMD as products or applications intended for one or more of the purposes listed under the definition of ‘medical device’, though they must be capable of operating without being part of any They must also feature the following characteristics: (a) they must be able to run on a general-purpose (i.e., non-medical) computing platform; (b) they can be coupled with other items (for example, as a module) or interact with other products.
- Nanomaterials: Natural, incidental, or manufactured materials containing particles in an unalloyed state, or in the form of an aggregate or agglomerate, where one or more of the external dimensions of at least 50% of the particles measure between 1 and 100 nanometers. Nanomaterials may include (a) fullerenes, graphene flakes, and single-walled carbon nanotubes with one or more external dimensions less than 1 nm; (b) manufactured materials with dimensions of up to 1000 nanometers that exceed the established limits of the nanoscale (between 1 and 100 nanometers), and which have size-dependent properties or phenomena that differ from the same material with dimensions on the macroscale.
- System: Defined as a set of compatible medical devices that relate to or interact with each other solely to achieve the purposes intended by the manufacturer (i.e., the device’s intended use).
Registration procedures and control frameworks
Resolution No. 751/2022 consolidates the requirements for the two frameworks used to regularize SaMD – ‘notification’ for class I and II products, and ‘marketing authorization’ for class III and IV products – as well as requirements for altering them. It establishes that SaMD must be classified according to their intrinsic risk to the health of users, patients, operators, or third parties – summarized as follows:
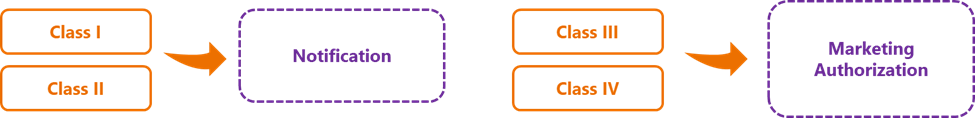
The risk classification system is based on 22 rules that consider the functionality, purpose and action mechanisms of these devices. One of the main updates concerns the specific rules for SaMD, nanomaterials, and active therapeutic devices with built-in diagnostic functionality for patient management.
Now that Resolution No. 751/2022 has taken effect, companies holding either of the two registrations for their products need to be aware that certain products may have new classifications and may fall under the opposite regularization framework. As such, the resolution has set specific deadlines to adjust the registration of the affected products.
Technical dossiers
The resolution has also created a new model for the table of contents in medical devices’ technical dossiers. This makes it possible to use dossiers prepared for foreign jurisdictions that comply with the International Medical Device Regulators Forum (IMDR) guidelines, of which Anvisa is a member. However, the previously established structure for technical dossiers remains accepted for products that were registered while Resolution No. 185/2001 was still effective, unless an application is submitted to change the registration.
Medical device documentation repository
In 2020, Anvisa created an online repository for storing and accessing digital documentation for medical devices. With Resolution No. 751/2022, registration holders are now required to add their medical device user instructions to the repository, and must ensure this information is kept up-to-date.
From now on, the owners of newly registered medical products (under either framework) or previously registered products that undergo changes must ensure the product’s registration application and instructions are added to Anvisa’s online repository within 30 days of these being published in Brazil’s Official Federal Gazette (DOU). As for any non-reportable changes that affect the user instructions, the registration application and instructions must be uploaded within 180 days of such changes being implemented.
Finally, it is important to note that any public advertising or communication regarding medical devices must be consistent with the information submitted by the product’s Anvisa registration holder.
For further information on medical devices, please contact Mattos Filho’s Life Sciences & Healthcare practice area.

